Quick Overview
SCRIBE (Single-Cell RNA-Seq Inference using Bayesian Estimation) takes a fundamentally different approach to single-cell RNA sequencing analysis. Instead of treating cells as isolated data points that need to be normalized, batch-corrected, and processed, SCRIBE views each cell as a sample from a statistical model that describes the entire dataset.
The Power of Probabilistic Modeling
At its core, SCRIBE embraces the inherent uncertainty in scRNA-seq data through Bayesian modeling. Consider a typical dataset with 10,000 cells and 20,000 genes —that’s 200 million data points! Rather than making point estimates about expression levels, SCRIBE learns probability distributions that capture:
How variable each gene’s expression truly is
Which zeros represent technical dropouts versus biological absence
How capture efficiency varies between cells
Whether cells belong to distinct subpopulations
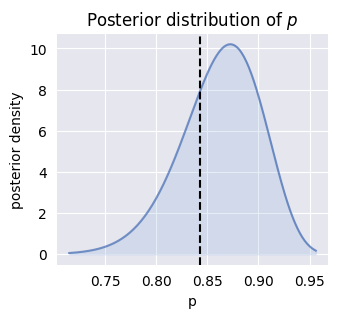
Rather than single values, SCRIBE learns complete probability distributions for model parameters. Here, the posterior distribution for the success probability parameter shows the range of plausible values given the data.
A Unified Framework for Inference
With high-dimensional data, no single inference method is perfect. SCRIBE provides three powerful, complementary methods accessible through a single, unified interface:
Stochastic Variational Inference (SVI): An optimization-based method that is extremely fast and scalable, perfect for exploring large datasets quickly.
Markov Chain Monte Carlo (MCMC): A sampling-based method that provides the gold-standard, exact posterior distribution for rigorous, publication-quality analysis. This requires specialized hardware such as GPUs with double precision support and a lot of memory (e.g., NVIDIA H100).
Variational Autoencoders (VAE): A deep learning approach that combines fast inference with powerful representation learning, ideal for tasks like dimensionality reduction and discovering latent structure.
This unified framework allows you to easily switch between speed and accuracy, choosing the right tool for the right stage of your analysis.
What Can You Do with SCRIBE?
Once SCRIBE learns your model, you can:
Generate normalized expression values with principled uncertainty estimates
Identify technical artifacts and batch effects probabilistically
Find cell subpopulations without arbitrary clustering
Make predictions about new cells
Compare different models to understand your data’s structure
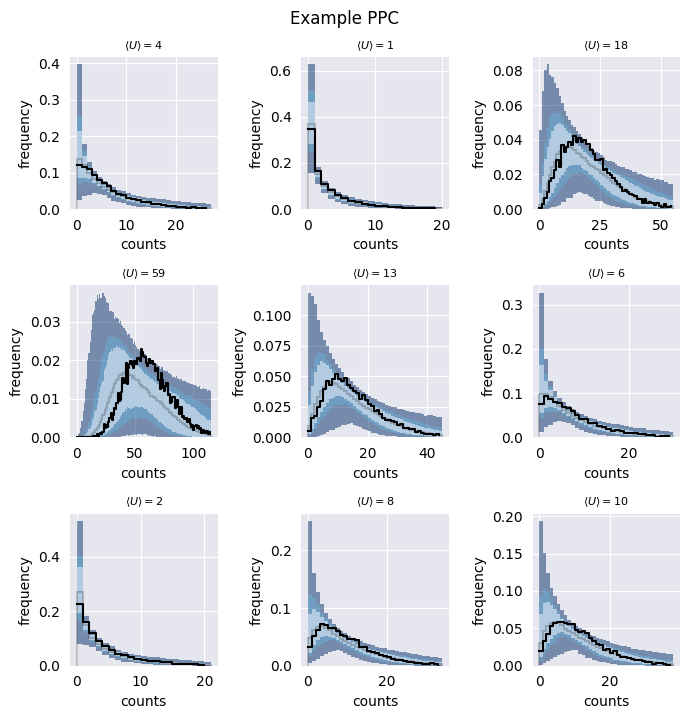
SCRIBE can generate synthetic data (blue bands) that matches the statistical properties of your real data (black line), allowing you to validate model fit and make predictions.
The Bayesian Advantage
The Bayesian framework provides several key benefits for single-cell analysis:
Specialized Models: Choose from models tailored to scRNA-seq data, such as the Zero-Inflated Negative Binomial (ZINB) and models that account for Variable Capture Probability (VCP).
Uncertainty Quantification: Every estimate comes with credible regions.
Model Comparison: Rigorously choose between competing biological hypotheses.
Principled Handling of Zeros: Distinguish between technical dropouts and true biological absence.
Integration: A natural framework for combining data from multiple experiments (to be developed).
Prediction: Generate realistic synthetic data for model validation.
SCRIBE makes these powerful Bayesian methods accessible through a simple Python interface, while maintaining the mathematical rigor necessary for proper statistical inference. Whether you’re interested in basic normalization or complex mixture modeling, SCRIBE provides a principled foundation for your single-cell analysis.